Miniconda3环境安装 -> 质控 -> 组装 -> 预测 -> 提取单拷贝直系同源基因 -> 建树
1. 前言 用4个或者多个片段构建的系统发育树节点支持率不高,以致于很多的类群分支无法解析。为了解决这一问题,在植物方面用了ddRAD、浅层基因组测序、基因组重测序、转录组测序等各种测序方法,但是分析方法主要采用提取SNP、组装叶绿体或转录组提取单拷贝直系同源基因。目前,从浅层基因组测序提取单拷贝直系同源基因的研究我一个都没有看到,所以也是尝试着摸了一下这种方法,期望能得到高的节点支持率和稳定的拓扑结构。
从2020年11月12号开始就一直在摸索这个从浅层基因组测序结果到系统发育树构建的流程探索,断断续续整了很久很久,终于是在邦叔和钊哥的指导下走完了整个流程。
2. 材料和方法 2.1 材料的选取 之所以选择浅层基因组测序,有很大一部分原因就是因为它可以使用硅胶干燥的分子材料,而且真菌的基因组比较小,大概也就50-100Mb,测5g的数据量也有50-100$\times$了,足够基因组的组装了。三代测序和转录组测序都需要液氮冷冻的材料,目前我并没有保存,明年如果采集到了拟口蘑属的材料会送测三代和转录组;不过目前也就是直接用分子材料测二代了。
2.2 分析流程 每次送测得到的结果有rawdata和cleandata,可以直接用rawdata分析。
每份标本都有一个单独的文件夹,里面有两个文件,分别是正向和反向的测序结果。
然后就可以开始分析了。
此分析流程均基于Windows Subsystem for Linux (WSL)完成,后续更大量的数据分析需要服务器运行。
2.2.0 安装环境 miniconda3 一些常用的软件基本都可以用miniconda3来安装,非常实用。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 wget -c https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh chmod 777 Miniconda3-latest-Linux-x86_64.sh bash Miniconda3-latest-Linux-x86_64.sh cd miniconda3/binchmod 777 activate . ./activate conda list conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/ conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/ conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/ conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
2.2.1 质控 fastqc 查看测序质量的软件。
1 2 3 4 5 conda install fastqc fastqc --help fastqc -o fastqc -t 20 */*.fq.gz
fastqc常用参数介绍:
-o:输出的文件夹
-t:运行的线程数
最后加上文件名。
fastp 删除低质量序列的软件。
1 2 3 4 5 6 7 conda install fastp fastp -h fastp -i Ge1416_FDSW202317667-1r_1.fq.gz \ -o out.R1.fq.gz -I Ge1416_FDSW202317667-1r_2.fq.gz \ -O out.R2.fq.gz -w 16
fastp常用参数介绍:
-i:正向测序输入文件
-o:正向测序输出文件
-I:反向测序输入文件
-O:反向测序输出文件
-w:线程数,最多只能16线程
2.2.2 组装基因组 spades 基因组组装软件。
1 2 3 4 5 conda install spades spades -h spades -o spades_result -1 out.R1.fq.gz -2 out.R2.fq.gz -t 20
spades常用参数介绍:
-o:输出文件夹
-1:正向测序结果(这里使用质控后的序列)
-2:反向测序结果
-t:线程数
busco 基因组组装评估的软件。
1 2 3 4 5 6 7 8 9 10 11 conda create -n buscoEnv -c bioconda -c conda-forge busco=4.1.2 augustus=3.3.3 biopython python conda activate buscoEnv mkdir -p ~/database/BUSCO/ cd ~/database/BUSCO/wget https://busco-data.ezlab.org/v4/data/lineages/basidiomycota_odb10.2020-09-10.tar.gz tar -xzvf basidiomycota_odb10.2020-09-10.tar.gz busco -i ./spades_result/contigs.fasta -o busco -m geno -c 10 -l ~/database/BUSCO/basidiomycota_odb10
busco常用参数介绍:
-i:输入文件
-o:输出文件夹
-m:模式选择:
geno or genome, for genome assemblies (DNA)
tran or transcriptome, for transcriptome assemblies (DNA)
prot or proteins, for annotated gene sets (protein)
-c:cpu数
-l:之前参考数据集存放的文件夹
之后还可以对组装结果的质量画一个图
1 2 3 cd ~/miniconda3/envs/buscoEnv/bintime python3 generate_plot.py -wd /mnt/e/getOganelle/tricholomopsis/1.rawdata/Ge1416_FDSW202317667-1r/busco
generate_plot.py的参数:
-wd:为所有busco检测结果数据所在的文件夹。在这个文件夹中包含了所有标本的short_summary.specific.basidiomycota_odb10.busco2.txt,为了区分,可以把busco2改成标本序号short_summary.specific.basidiomycota_odb10.Ding384.txt,其中Ding384就是我一份标本的序号。评估结果图如下:
Assembly-stats 评估contigs和scaffolds长度的软件
1 2 3 4 5 6 7 conda install assembly-stats assembly-stats contigs.fasta >> assembly-stats.csv assembly-stats scaffolds.fasta >> assembly-stats2.csv conda deactivate
2.2.3 基因组的预测 RepeatModeler + RepeatMasker 屏蔽重复序列。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 conda install RepeatMasker conda install repeatmodeler BuildDatabase -name zhao2118 -engine ncbi spades_result/contigs.fasta RepeatModeler -database zhao2118 -engine ncbi -pa 10 &> zhao2118.out & nohup RepeatMasker -e rmblast -lib zhao2118-families.fa -pa 16 spades_result/contigs.fasta &
Augustus 从头预测(de nova预测)软件,需要使用屏蔽了重复序列的文件。
1 2 3 4 5 6 7 conda activate buscoEnv augustus --species=help augustus --species=coprinus_cinereus contigs.fasta.masked > test.gff
GenomaThreader 同源预测软件。
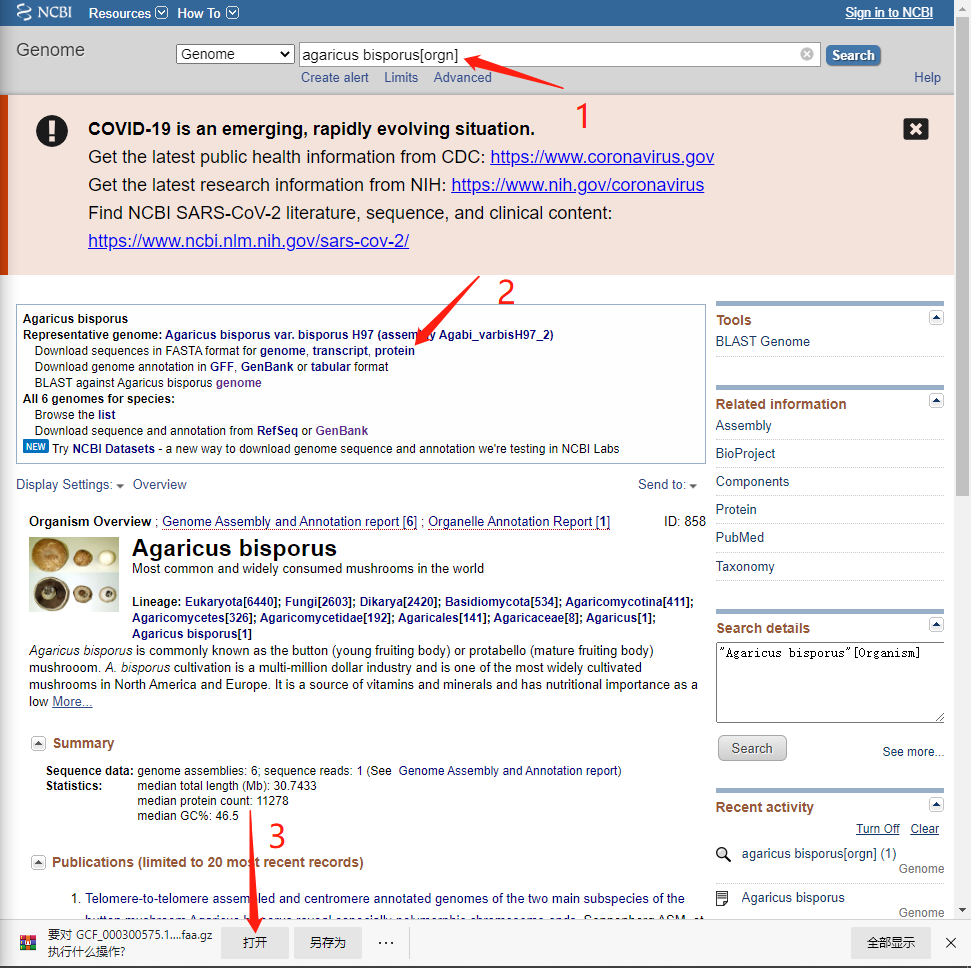
首先要在NCBI上下载近缘物种的蛋白注释序列,如下图。首先在Genome里面搜索需要的物种,以Agaricus bisporus 为例,然后点击”Download sequences in FASTA format for genome, transcript, protein”中的protein就可以下载到蛋白序列了。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 conda install genomethreader cat Armillaria_solidipes.faa\ Gymnopilus_dilepis.faa\ Mycena_chlorophos.faa\ Pterula_gracilis.faa\ Tricholoma_musutake.faa >all.pep.fa makeblastdb -in spades_result/contigs.fasta -parse_seqids -dbtype nucl -out index/ding384 tblastn -query ../all.pep.fa -out ding384.blast -db index/ding384 -outfmt 6 -evalue 1e-10 -num_threads 8 -qcov_hsp_perc 50.0 -num_alignments 5 awk '{print $1}' ding384.blast >ding384.list sudo apt install john sort ding384.list| uniq >ding384.ho.list conda install seqkit seqkit seq ../all.pep.fa -w 0 > all.fa cat ding384.ho.list | while read line;do grep "$line " -A 1 all.fa >$line .1; done ;cat *.1 > ding384.homo.fa; rm -rf *.1 gth -genomic spades_result/contigs.fasta -protein ding384.homo.fa -intermediate -gff3out > ding384_gth.gff grep -v "^#" ding384_gth.gff | grep -v "prime_cis_splice_site" | awk -F ";" '{print$1}' >ding384.homo.gff less ding384.homo.gff
钊哥师姐写的脚本,change-gff_lym.py
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 import sysimport refout = open (sys.argv[3 ],'w' ) ref_dict={} with open (sys.argv[1 ]) as gff: for line in gff: line_s = line.strip().split('\t' ) if 'gene' == line_s[2 ]: ref_g = re.split('=' ,line_s[8 ]) ref_gene = ref_g[1 ] ref_dict[ref_gene]=[] else : pos = line_s[3 ] ref_dict[ref_gene].append(pos) with open (sys.argv[2 ]) as gff_r: for eachline in gff_r: i = eachline.strip().split('\t' ) info = re.split('=' ,i[8 ]) name = info[1 ] ref_set = [] for n in range (0 ,8 ): ref_set.append(i[n]) ref_list = '\t' .join(ref_set) ref_set1 = ['CDS' if x == 'exon' else x for x in ref_set] ref_list1 = '\t' .join(ref_set1) if 'gene' == i[2 ]: fout.write(eachline) elif 'exon' == i[2 ]: if name in ref_dict: vs = ref_dict[name] len_vs = len (vs) for a in range (0 ,len_vs): if vs[a] == i[3 ]: b = vs.index(vs[a]) fout.write('%s\tID=%s.t%d;Parent=%s\n' %(ref_list,name,b+1 ,name)) fout.write('%s\tID=%s.c%d;Parent=%s\n' %(ref_list1,name,b+1 ,name)) fout.close()
1 python ../change-gff_lym.py ding384.homo.gff ding384.homo.gff prediction.ding384.gff
EvidenceModeler 整合预测结果。目前因为内存不足,无法运行该软件。暂时不写这部分的内容。
2.2.4 提取单拷贝直系同源基因 因为无法整合多个软件预测的结果,目前只能用augustus或gth预测的结果来提取,在这里我用了augustus的结果。
Gffread gff格式转换的软件,在提取单拷贝直系同源基因之前,我们需要用gffread把gff文件转换成fasta格式的文件。
1 2 3 4 5 6 7 8 conda install gffread gffread ge1416.gff -g ../Ge1416_FDSW202317667-1r/spades_result/contigs.fasta.masked -w Ge1416_extron.fasta -x Ge1416_cds.fasta -y Ge1416_protein.fasta mkdir protein cp *_protein.fasta protein cd protein
给每条序列改名字,加上标本编号
1 2 sed 's/>/>ge1416./g' Ge1416_protein.fasta > ge1416.fasta
改完名以后的序列。
Orthofinder 快速、准确和全面的比较基因组学分析工具。
1 2 3 4 conda install orthofinder nohup orthofinder -t 16 -f protein/ -S blast &
orthofinder常用参数介绍:
-t:线程数
-f:输入目录
-S:选择比对模式
如果要去掉某一物种,在SpeciesIDs.txt中将该物种注释掉
1 $nohup orthofinder -b WorkingDirectory
如果我们想增加额外的物种进行分析,可以按照如下方式运行
1 $nohup orthofinder -b WorkingDirectory -f new_fasta_directory
2.2.5 利用单拷贝知悉同源基因构建系统发育树 本来系统发育树的构建已经写过很多次了,但是发现用超多片段构建和少数片段构建的差异还是挺大的,也是摸索了好几天,还是记录一下吧。
Mafft 序列比对
1 2 3 4 5 6 7 8 9 wget https://mafft.cbrc.jp/alignment/software/mafft_7.475-1_amd64.deb sudo dpkg -i mafft_7.475-1_amd64.deb mafft -h for i in *.fa;do linsi ${i} > ${i} .mafft; done
mafft的使用可以看说明书,我个人的习惯是在每一步的后面加上这一步运行的软件名作为后缀
Gblocks 提取保守区域序列
1 2 3 4 5 6 7 8 9 10 11 12 wget http://molevol.cmima.csic.es/castresana/Gblocks/Gblocks_Linux64_0.91b.tar.Z sudo apt-get install -y ncompress tar Zxf Gblocks_Linux64_0.91b.tar.Z echo 'PATH=$PATH:~/Gblocks_0.91b/' >> ~/.bashrcsource ~/.bashrcGblocks -h for i in *.mafft;do Gblocks $i -b4=2 -b5=h -t=p -e=.gblocks; done
Seqkit 这个是序列包,可以花式处理序列,非常好用。
1 2 3 4 5 6 7 8 9 10 11 conda install seqkit for i in *.mafft-gb;do seqkit sort $i > $i .sort; done for i in $.sort;do seqkit seq $i -w 0 > $i .seq; done