NGS20220312
NGS 分析流程
首先,拿到的数据。
0. 去PCR重复(可选)
Fastunique
1. 数据质控(可选)
1 | fastqc -o fastqc -t 20 Ge1416_FDSW202317667-1r_R1.fq.gz |
输出的文件: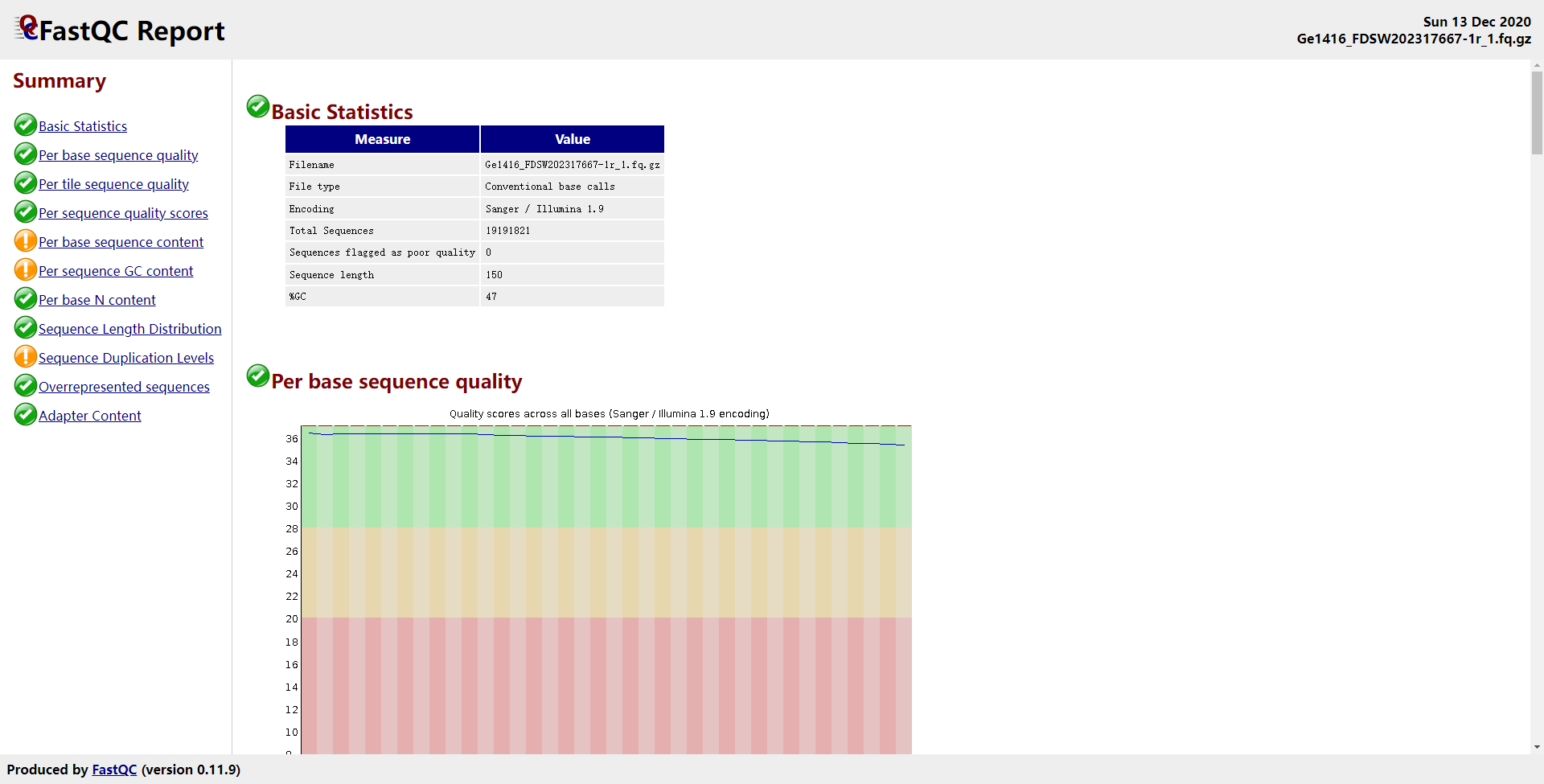
2. 数据清洗
1 | fastp -i Ge1416_FDSW202317667-1r_R1.fq.gz -o Gr1416_R1.fq.gz -I Ge1416_FDSW202317667-1r_R2.fq.gz -O Ge1416_R2.fq.gz; |
输出文件:
Ge1416.R1.fq.gz
Ge1416.R2.fq.gz
3. 组装基因组
1 | spades -o spades_result -1 Ge1416.R1.fq.gz -2 Ge1416.R2.fq.gz -t 20 |
输出一个文件夹: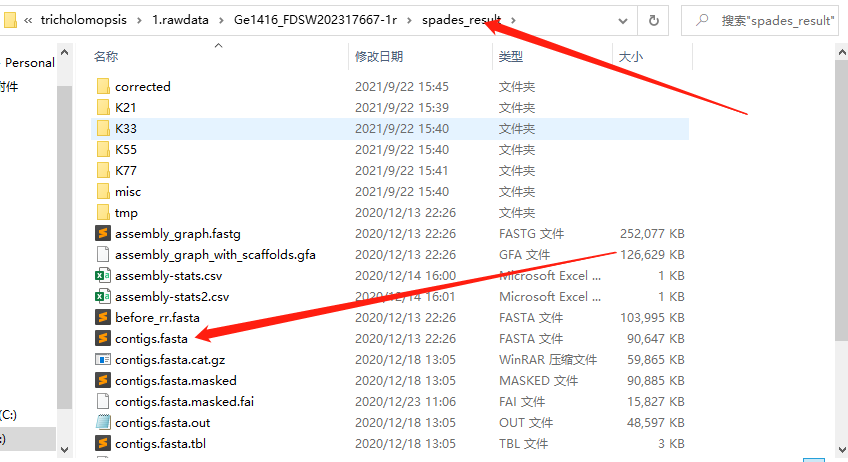
有用的文件是:
contigs.fasta
scaffords.fasta
3.1 去除unpaired序列
这里我选用了trimmomatic来执行
1 | # 软件安装 |
在RED文件下中运行命令:
5. 注释
1 | augustus --species=coprinus_cinereus contigs.msk > Ge1416.gff |
6. 提取proteins
1 | gffread Ge1416.gff -g contigs.msk |
7. 提取BUSCO
1 | busco -i Megacollybia_platyphylla.fna -o Megacollybia_platyphylla -m geno -c 20 -l ~/database/BUSCO/agaricales_odb10 |
在这个文件夹下面就是单拷贝直系同源基因(BUSCOs)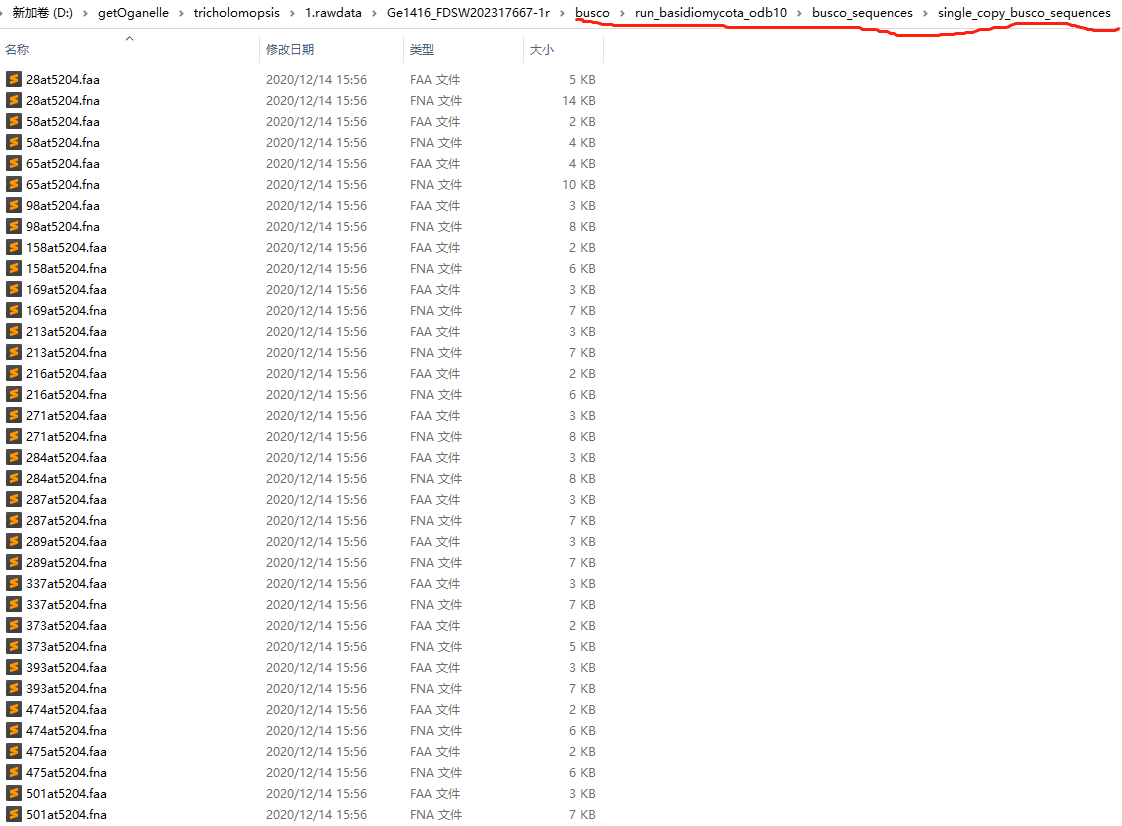
不同的标本,提取的数量是不一样的
7.1 质量作图
python3 ~/miniconda3/envs/buscoEnv/bin/generate_plot.py -wd /mnt/d/Agaricales5/genomequality
——-分界线——-
开始建树的过程
运行完上面的过程,我们可以得到每个标本都有自己的一个文件夹,
![[Pasted image 20220102173628.png]]
在路径\run_agaricales_odb10\busco_sequences\single_copy_busco_sequences,就是我们要用的BUSCOs。
8. 基因数量统计
8.1 定制一个统计基因组大小的循环
1 | dos2unix list1.txt |
8.2统计每个基因在多少个物种中存在
1 | dos2unix list2.txt |
list2.txt中,储存了3868个基因的名字。
手动打开genespe.txt,把其中要用的基因的名字复制到一个文件中,例如“gene200.txt”。
8.3 把所有物种的每个基因单独放入一个文件中
1 | dos2unix list3.txt |
其中,list.txt是所有物种的名单,list2.txt是3870个基因的名单,list3.txt是挑选出来的基因名单。
1 | for i in $list;do |
9. 序列比对
1 | for i in *.fasta;do |
10. 提取保守序列
1 | for i in *.mafft;do |
11. 序列合并
1 | for i in *.mafft-gb;do |
12. 建树!
1 | raxmlHPC-PTHREADS-SSE3 -T 20 -f a -x 123 -p 123 -N 1000 -m PROTGAMMAIJTTF -k -O -n all.tre -s all.fa |
今天发现其实建树的话,组装完基因组直接用BUSCO寻找单拷贝直系同源基因就可以了,不需要对基因组进行注释,这样的话就省了很多步骤。
问题&概念
baits:这个应该如何理解,我并不是特别明白。
CDS:coding sequences
