MCMCtree 计算分化时间
目前我知道的可以做物种分化时间计算的软件主要有beast,beast2,r8s,paml的mcmctree,还有Nagy论文中的PhyloBayes (R package)。目前呢,我是尝试使用了beast2,非常的慢,我84个物种接近50万AA (amino acids) 每100万代运算时间差不多27h,但是一般设置是1亿代也就是需要100多天,年都过完了。暂且不提,所以开始尝试MCMCtree和r8s,本篇就是mcmctree的使用记录
1. 安装
本文主要介绍在Linux或者WSL上的安装,直接根据官网的教程走就可以了
1 | # 下载 |
2. 文件的准备
网上常见的教程基本都在说MCMCtree用碱基序列来估算时间序列比较方便,陈连福也说了碱基序列更具有可靠性(这里我不是很理解,为什么碱基序列更具有可靠性),但是氨基酸建树的教程也有,在官方文档里边Toturial 4也是氨基酸的教程,主要参考的就是这俩。
需要准备三个文件
2.1 有根树的文件,不需要枝长,需要添加化石节点,化石节点的话是在相应的位置打个单引号,单位是MYA,也就是百万年,比如105个百万年到210个百万年,就是$’>1.05<2.10’$。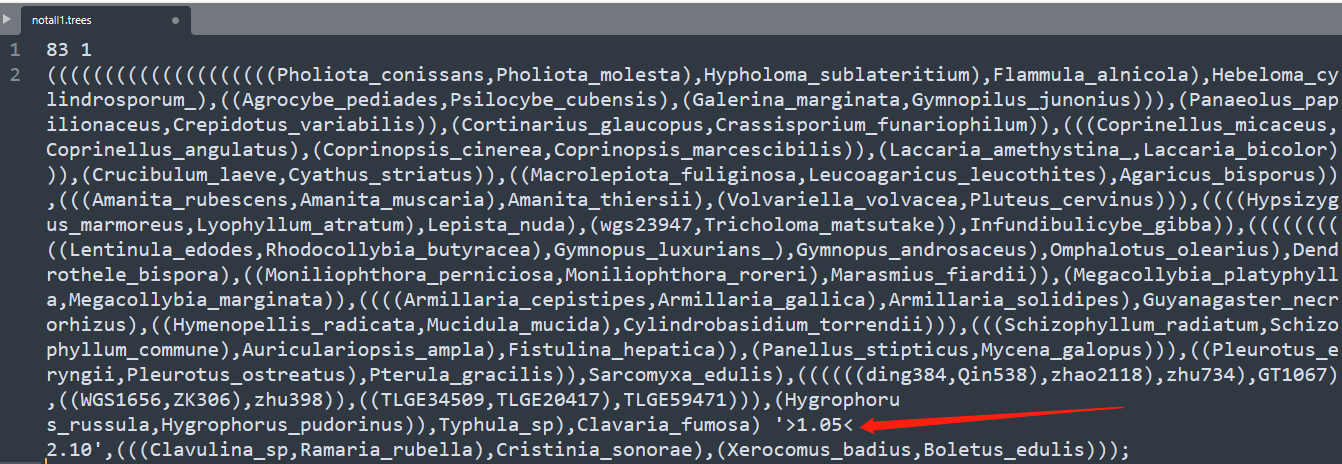
这个文件可以通过RaxML构建的树bipartition提取出来,我不知道这么提取出来没有枝长的树,所以是提取了文件以后输入:
1 | sed "s/\:[0-9\.]//g" all.trees > all1.trees |
2.2 序列文件,需要时phylip格式的
这一步很多都可以转,比如我有一个从钊哥那里继承的祖传代码:fas2phy.py
1 | import re |
或者用R语言来弄:
1 | library(devtools) |
然后因为有些序列之中包含了奇怪的氨基酸序列,比如B,U,这两个建议直接手动替换成X或者-,删除会缩减序列长度。因为肯定不会很多,批量删除的话容易把序列名称中的B和U改掉。
2.3 mcmctree.ctl文件
建议的是照着图中标红的区域对文件进行更改,其他的不建议动
mcmctree.ctl
1 | seed = -1 |
3. 运行程序
基本上完成上面三个文件的准备就需要
1 | # 最后使用 |
在这里会发生一个报错: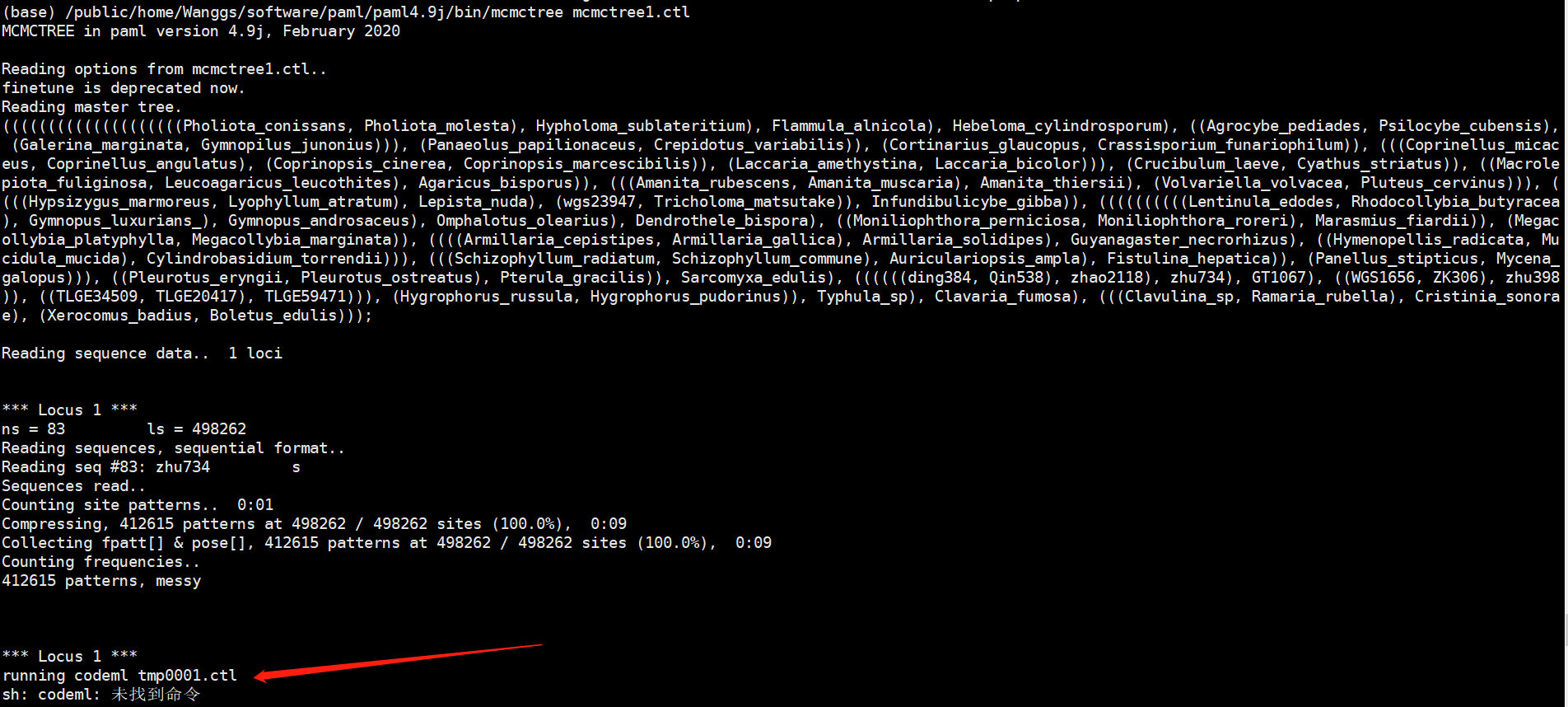
这个是正常的,如下是官方教程所示:
我们需要手动的把tmp0001.ctl文件按照图中所示进行更改,删掉“out.BV”和“rst”两个文件,并且把“wag.dat“从dat文件夹中拷贝到这个文件夹里就可以执行下一步了。
1 | codeml tmp0001.ctl |
这样就使用WAG+Gamma生成了适当的Hessian矩阵,接下来将rst2重命名为in.BV,现在可以更改mcmctree.ctl 的 usedata = 2
回到上一目录,新建一个文件夹,将 那三个文件和新生成的in.BV拷贝进去
接下来运行
1 | mcmctree mcmctree.ctl |
4. 补充
4.1 一条龙服务
整体来讲,按上述内容走就可以很好的运行了,但我还是觉得非常之麻烦,于是写了一个mcmctree.sh
1 | mcmctree mcmctree.ctl; |
就是我们在一个文件夹中放入蛋白序列文件(*.aa)、树文件(*.trees)和mcmctree.ctl文件,并创建一个step3文件夹,把这三个文件复制进去,并在这个文件夹的上一级文件夹中放入wag.dat和tmp0001.ctl就可以直接运行mcmctree.sh了。
4.2 关于收敛
为了让数据收敛其实也是一个非常困难的事情,可以根据参考资料中提供的信息来设置运算代数。

在大量数据的时候我的数据验证结果为:burnin 2000k; sampfreq 10; nsample 1000k会更好一点。
于此,mcmctree和r8s计算分化时间的全部工作就结束了!
参考资料:
PAML的官方网站:
Phylogenetic Analysis by Maximum Likelihood (PAML) (ucl.ac.uk)
这一篇介绍了原理:
估算系统树分歧时间 —— paml.mcmctree,r8s | 生信技工
(yanzhongsino.github.io)
这几篇是介绍了MCMC估算的pepline:
mcmctree 定年 —— 使用氨基酸序列 (gitee.io)
mcmctree估算物种分歧时间 - 简书 (jianshu.com)
使用PAML进行分歧时间计算 | 陈连福的生信博客 (chenlianfu.com)
这一篇介绍了很多参数
PAML软件中的mcmctree命令估算物种分歧时间 - BPSO_mynotes - 博客园 (cnblogs.com)
这一篇介绍的比较全面,但是都没讲清楚:
【比较基因组】从进化树到分化时间 - 简书 (jianshu.com)
这一篇是介绍r8s的:
使用 r8s 估算物种分歧时间 | 陈连福的生信博客 (chenlianfu.com)
